Benchmark and train machine learned interatomic potentials within atomistic modeling
Master projects/internships - Leuven | Just now
Systematic benchmark and train of Machine Learning interatomic potentials for studying amorphous materials and complex interfaces
The age of artificial intelligence (AI) has begun, leading to its extensive application across numerous fields. The AI models currently in use are built on advanced semiconductor chips (CMOS) that utilize the latest fabrication technologies and methodologies, resulting in enhanced performance and reduced costs. As the quest for improved performance in CMOS devices continues, there is a growing necessity for new materials that can facilitate the miniaturization of these components.
Progress in this area depends on a comprehensive understanding of the fundamental properties that govern complex material interfaces, along with the establishment of effective design principles for devices. Materials modeling approaches are employed to gain a thorough understanding of fundamental properties. These simulations can be broadly classified into two categories: the first is based on quantum mechanical methods, such as density functional theory, and the second relies on semiempirical analytical interatomic potentials or force fields. Quantum mechanical methods (first-principles or ab initio) offer both versatility and accuracy, but they are computationally intensive. In contrast, more cost-effective solutions can be achieved through semiempirical methods, which simplify the representation of interatomic interactions using parameterized analytical functional forms.
A new class of interatomic potentials, known as machine learning potentials (MLP), has been developed to enhance accuracy comparable to ab initio methods while significantly reducing computational demands. These MLPs are trained on extensive datasets derived from hundreds of thousands of ab initio simulations of bulk crystalline materials. However, their effectiveness in modeling complex interfaces and amorphous materials remains unverified. This proposal aims to address this gap by establishing a systematic benchmark for existing amorphous materials and complex interfaces, as well as by retraining these MLPs for specific applications in modeling complex structures.
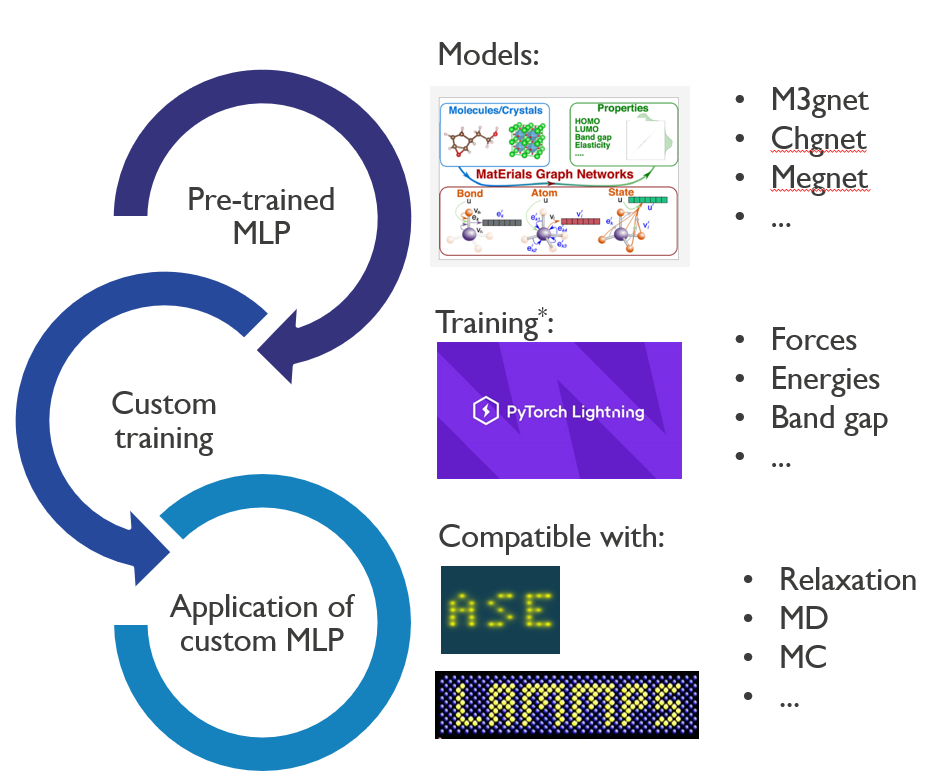
You will collaborate with experts in atomistic modeling and have access to a substantial computational infrastructure equipped with CPUs and GPUs, which are utilized for a broad range of computational modeling activities at Imec. We are looking for a self-motivated individual who is a team player and possesses knowledge of machine learning and Python in a Unix environment. Familiarity with ab initio simulations is an added advantage.
Type of Project: Combination of internship and thesis
Master's degree: Master of Science
Master program: Chemistry/Chemical Engineering; Computer Science; Materials Engineering; Nanoscience & Nanotechnology; Physics
Duration: 4 months
Supervisor: Michel Houssa
For more information or application, please contact the supervising scientist Kiroubanand Sankaran (Kiroubanand.Sankaran@imec.be).
Only for self-supporting students.
![]()